For Alzheimer’s patients and their care providers, amyloid beta is memory enemy number one. While all brains make this small molecule, the brains of Alzheimer’s patients seem unable to get rid of it. However, James Keaney, a postdoc at Trinity College in Dublin, can coax amyloid beta out of a mouse’s brain through its blood vessels.
That’s no easy task—in mammals, the brain’s blood vessels “have developed an extra protection, what we call the blood brain barrier,” Keaney said. Normally, the barrier only lets desirable substances through and keeps intruders out of the delicate brain tissue.
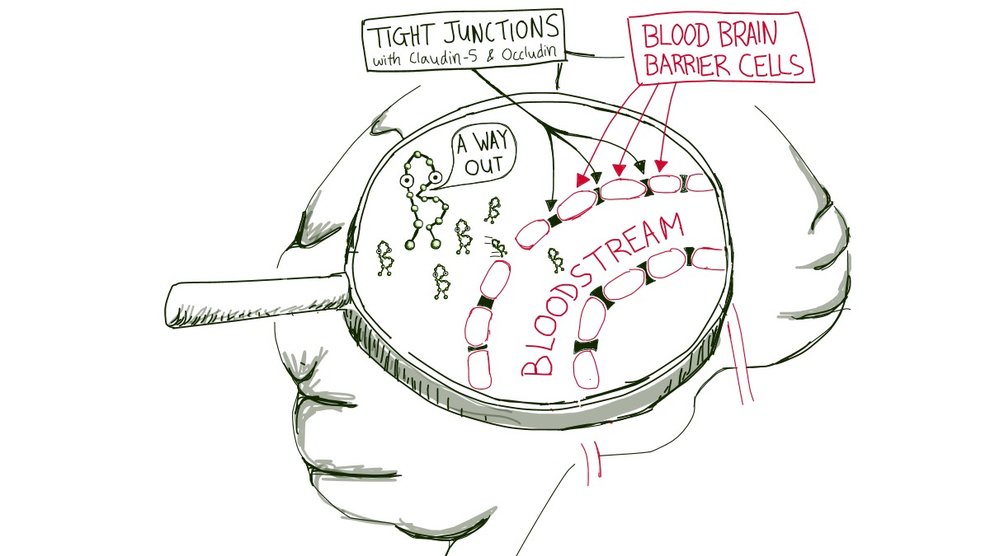
But he can make the barrier leakier if he lowers the levels of two proteins: claudin-5 and occludin, which are key components of the tight junctions that stitch individual barrier cells into a single, unified wall. After lowering the levels of claudin-5 and occludin in the tight junctions of mice, Keaney could detect even higher levels of amyloid beta in their blood, indicating an even leakier blood brain barrier.
Keaney’s team also found that healthy brains might use the same strategy to prevent a build-up of amyloid beta. When they exposed blood brain barrier cells in a petri dish to high levels of amyloid beta, the cells decreased their claudin-5 and occludin levels by themselves, implying that healthy mouse brains might temporarily increase the leakiness of their barrier when there is too much amyloid beta. If mouse brains do this, ours may, too.

In fact, Keaney also has some evidence to suggest that our brains use a similar mechanism. Since experimenting on live humans’ brains is too risky, he conducted autopsies on deceased Alzheimer’s patients. As predicted, the levels of claudin-5 and occludin were very low, suggesting that their brains were “trying” to clear the excess amyloid beta, he said. But there was still so much amyloid beta that the toxin had formed large conglomerations around the vessels called plaques—a telltale sign of advanced Alzheimer’s.
Why didn’t the leak-it-out mechanism work for these people? “Plaques are really sticky,” Keaney said. “They can’t move and they’re difficult to break up. At this late stage in Alzheimer’s, it’s too large to pass between cells anymore.”
If this line of research is to lead to a treatment, scientists must also find ways to lower the levels of claudin-5 and occludin early in life before the amyloid beta accumulates. Even if they do find a way, such a dramatic change to brain security is always risky. That security is there for a reason, Keaney said. “You almost never hear of someone getting a brain infection.”
Such treatments would also require a better understanding of the various states of the amyloid beta molecule itself, which is cut “from a larger precursor protein that exists in the membranes of all neurons,” Keaney said. This freshly cut amyloid beta can then transition into other forms, one of them being plaques.
But a 2013 study in Nature by researchers at Korea University, demonstrated that brain cells in culture were largely unharmed by amyloid beta plaques. An MIT post-doc (who wishes to stay anonymous) pointed out that there is “accumulating evidence that the short-lived transition from monomer to plaque is the neurotoxic species,” rather than the plaque form itself. Researchers must identify what forms of amyloid beta are causing the neurological damage associated with Alzheimer’s in order to know if Keaney and his team are on the path to a viable treatment.
